研究テーマ¶
第一原理熱力学計算法の開発と応用¶
量子力学の基本原理から出発し,物理現象を非経験的に記述する手法を第一原理法(first principles methods)と呼びます.第一原理法を材料研究に応用する試みは1980年代に始まり,その後,計算機と計算技術の進歩により着実に成果を上げてきました.そして21世紀に入ったころ,さらに大きな進歩を遂げました.温度の効果を定量的に計算できるようになったのです.その結果,物質の比熱や自由エネルギーのような熱力学関数が第一原理計算で求められるようになりました.そして状態図や相転移,拡散のような材料科学の根幹となる現象に対し,アプローチできるようになったのです.これにより第一原理計算の守備範囲は,物質設計から,材料を合成するためのプロセス設計まで大きく拡がりました.このような研究は,世界中でまだ端緒についたばかりですが,当研究室では,酸化物セラミックス材料や半導体,金属材料において,すでに多くの先駆的な成果を報告しています.
第一原理熱力学計算では,従来の第一原理計算と,様々な新しい手法とを組み合わせます.格子振動の効果を精確に求めるためには,フォノンの状態密度を第一原理法で求めます.固溶体や化合物の原子配列を議論するためには,クラスター展開法やモンテカルロ法などの統計力学手法と第一原理法を組み合わせます.拡散現象を扱うには,原子ジャンプの素過程を第一原理法で定量化します.当グループでは,このような新しい計算方法を材料科学の問題に積極的に応用しています.
第一原理熱力学計算によるZnO-MgO擬2元系状態図
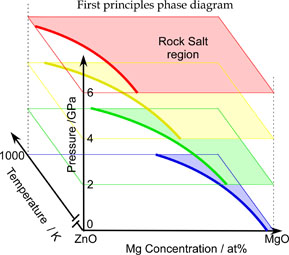
予測されたZnO-MgO相境界の温度及び圧力依存性は,実験結果を良く再現します.第一原理に基づいたこの状態図計算手法は実験が困難な系にも適用でき,材料の設計及び開発における重要なツールとなります.
ZrO2の相対自由エネルギーの第一原理熱力学計算
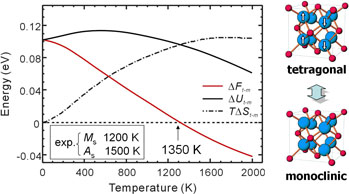
monoclinic相とtetragonal相の自由エネルギー差から求めた相転移温度1350Kは,報告されている実験結果と良く一致しています.
第一原理計算に基づいたマテリアルズ・インフォマティクスの展開¶
高速で大容量化した計算機を利用して膨大な情報を統合・整理したうえで,必要な知識を取り出すデータマイニングと呼ばれる技法が,自然科学や社会科学の多様な分野で活用されています.自然科学への応用の代表例がバイオ・インフォマティクスと呼ばれる生命科学と情報科学との融合領域です.これは遺伝子に関する膨大な実験データを計算機で解析し,その情報に基づいて生命現象を理解するというもので,すでに大きな成果を上げています.同様に,計算機の巨大情報処理能力を材料研究に応用する学術領域を,私たちはマテリアルズ・インフォマティクス(材料インフォマティクス)と呼んでいます.材料に関する構造や特性など様々な情報をデータベースとして集約し,それを適切に整理することで,材料設計・探索に利用するのです. 最近,計算機の性能向上と効率的な計算手法の出現,そして計算精度が大幅に向上したことで,第一原理計算を多数実行し,系統的に均質な物質情報を獲得し,データベース化することが可能になってきました.しかし,データベースを構築するだけでは,材料設計・探索には全く不十分です.様々なスケールでの階層構造の性質が創発的に現れている材料機能を物質の一次情報に基づいて合理的に推定すること.そして多元系における化合物の巨大な自由度のなかから,最適組成をハイスループット・スクリーニングすることが必要なのです.当グループでは,このための新しい技術開発を進めており,様々な材料系を対象として材料設計・探索への応用を目指しています.
第一原理計算に基づいたマテリアルズ・インフォマティクスの模式図

リンク
第一原理計算に基づいたマテリアルズ・インフォマティクス
[PDF]
新しい結晶構造と機能の探索¶
構成元素数や組成が単純である無機化合物の多くは,すでに結晶構造が決定され,その物性値も知られています.しかし,高圧下や気相成長法などの準安定状態で合成される化合物は,未知な構造や物性の宝庫です.当グループでは,典型元素酸化物の高圧相や結晶多形,相転移挙動について第一原理計算を応用した系統的な研究を行っているほか,実験的に未知であった不定比酸化物について結晶構造決定や相安定性を明らかにしました.
このような研究の中で,これまでの強誘電体材料とは異なるメカニズムで分極挙動を示す一連の複合酸化物材料を発見したほか,多くの新規高圧相の構造を明らかにしました.
LaAlO3の結晶構造の圧力依存性
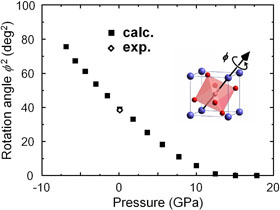
負荷圧力約13GPaでAlO6八面体の回転角がゼロとなり,rhombohedral相からcubic相に転移することが予測されます.この結果はフォノンの計算からも確認され,最近報告された実験結果を良く再現しています.
XANESとELNESのナノ材料科学への応用¶
X線吸収スペクトルの吸収端近傍微細構造(XANES)は,内殻軌道から非占有軌道への電子励起に起因することから,注目する材料のナノ構造を反映します.放射光施設を利用して測定した場合,実験方法を工夫すれば,ppmオーダーの不純物や,材料の表面,界面を修飾している不純物の局所環境を評価することができます.これと同様の情報を与える分光法に,電子顕微鏡を利用した電子エネルギー損失分光励起端近傍微細構造(ELNES)法があります.この手法は,X線を利用した実験と異なり,微細組織と対応させた状態分析ができるというユニークな特長を持ちます.最先端の電子顕微鏡に装備した電子線エネルギー損失分光(EELS)装置を用いれば,電子ビーム径0.1nmでの原子カラム毎の測定も可能です.
このような電子分光の結果は,これまで,参照物質のfinger print(指紋)と照合することによって解釈されてきました.しかし指紋照合法は参照物質がない場合に適用できません.また指紋が大雑把に合致しても,状態を同定するための必要条件を満足するだけであり,十分条件とはなりません.電子分光の理論スペクトルの適切な計算手段があれば,参照物質がなくとも実験結果を局所原子配列や化学結合状態といった情報に変換することが可能です.当グループでは,このXANESやELNESの計算手法を開発すると同時に放射光や電子顕微鏡を用いた実験を行い,これらの分析法を材料科学の様々な問題に適用すべく研究を進めています.
XANES及びELNESの概念図
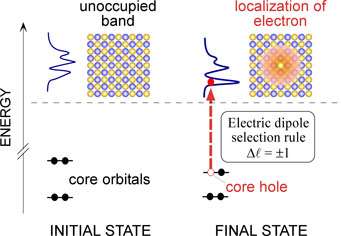
XANES及びELNESを与える励起過程では,励起先の非占有軌道は内殻に残された正孔(コアホール)近傍に局在した状態となります.本研究室では,この効果を取り入れたXANES及びELNES理論スペクトルの計算手法を開発しています.
MnO及びMn添加ZnOからのMn L2,3端XANESとその第一原理多電子計算
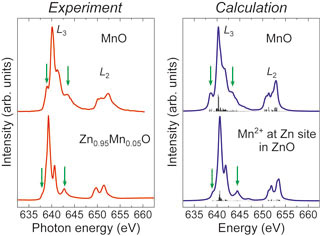
多電子論に基づいた新しい第一原理計算法を開発し,これをMn L2,3端XANESの解釈に適用した結果,ZnO中のMnドーパントが2価のハイスピン状態でZn位置に置換固溶していることがわかりました.
リチウムイオン2次電池の材料科学¶
現在わが国では,年産10億個近いリチウムイオン2次電池が生産され,携帯電話,ノートパソコンなどに広く用いられています.その製造技術はすでに成熟したものですが,その一方で,近年相次いで発火事故を起こしていることからも分かるように,安全・安心の観点から十分なものとはなっていません.それにもかかわらず電池の高容量化,高性能化への要求も依然として高く,さらにハイブリッド型自動車にリチウムイオン2次電池を応用しようという研究も精力的に進められています.
当グループでは,装置としての電池の開発研究を直接行うことはありませんが,第一原理計算と電子分光手法による評価を併用して,電池材料における固体化学反応の素過程を解明し,新しい材料開発に繋げることを目指しています.これまでに,新しい正極材料の設計,負極グラファイト中のリチウム拡散のシミュレーション,電池の全固体化に向けた固体電解質材料の設計など多くの先駆的な成果を上げてきました.
LiC6中のLiの移動経路とエネルギープロファイル
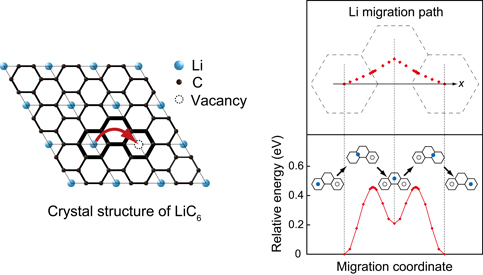
充放電過程において負極グラファイトへのリチウム層間挿入により形成されるLiC6中で,Liは準安定な中間状態を経た2段階のジャンプにより移動することが予測されました.このモデルに基づいて算出した拡散係数は,報告されている実験値とよく一致します.