機械学習を活用した結晶構造探索手法の開発と応用¶
機械学習ポテンシャルの大域的構造探索への応用
機械学習ポテンシャルは、材料科学における大域的構造探索において非常に有効な手法です。最近では、第一原理計算や遺伝的アルゴリズムを組み合わせた構造探索が手軽に利用できるようになっていますが、特に構造が未知の多元系や高圧相、また原子数が多い単位胞の系においては、真の安定構造を見つけることが依然として難しいという課題があります。その主な理由は、原子数が増えると最適化の自由度が指数関数的に増加し、安定構造の探索が複雑になるためです。 それでも、機械学習ポテンシャルを活用することで、膨大な数の結晶構造に対して効率的にエネルギー計算を行い、化学組成とポテンシャルに基づく広範囲な構造探索が可能になります。機械学習ポテンシャルは第一原理計算に基づいて構築されているため、その精度を保ちながら計算効率を大幅に向上させることができます。このアプローチにより、準安定構造の列挙や大規模な構造探索が実現し、より効率的な新しい材料の発見が進むことが期待されています。
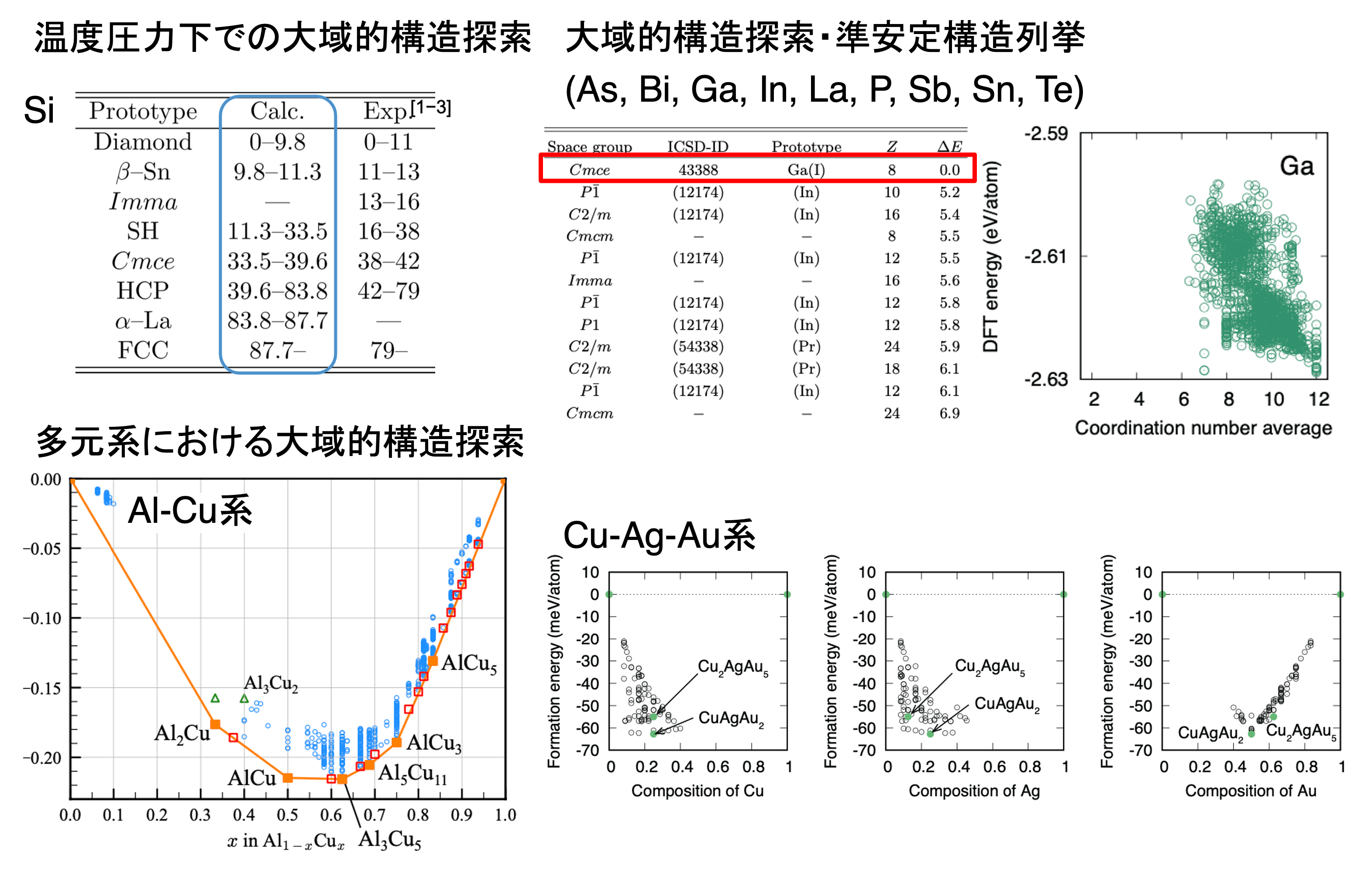
機械学習ポテンシャルによる大域的結晶構造探索 [A. Seko, PRB (2024), A. Seko, arXiv:2407.20630 (2024), H. Wakai, A. Seko, I. Tanaka, J. Ceram. Soc. Japan (2023)]
最適化手法やアルゴリズム手法の結晶構造探索への応用
結晶構造探索では、事前知識や仮定を元に離散的な構造候補集合を列挙し、その中から最適な構造を選択する手法が広く用いられています。特に、多成分系における置換型結晶構造の探索では、格子モデルを仮定し、原子配置を列挙して解を導く方法が一般的です。このアプローチにより、連続的な探索空間に比べてより広範囲な構造探索が可能になります。 しかし、候補となる構造集合が非常に大きくなると、すべての候補に対して第一原理計算を実行することは現実的ではなく、効率的な方法が必要になります。ここで機械学習を活用することで、計算コストを抑えつつ、探索の効率を大幅に向上させることができます。
例えば、酸素欠損ペロブスカイトの構造予測では、まず膨大な数の非等価な酸素欠損配置を列挙し、約400~500個の構造をサンプリングして第一原理計算を実行しました。その後、構造特徴量として酸素サイトの対の相関関数を用いてエネルギーモデルを構築し、ガウス過程回帰を使って予測しました。予測誤差が大きい場合にはベイズ最適化を適用し、逐次的に最適な構造を探索しました。この手法により、既存の酸素欠損ペロブスカイト構造に加え、新たな規則的酸素欠損構造が予測されました。
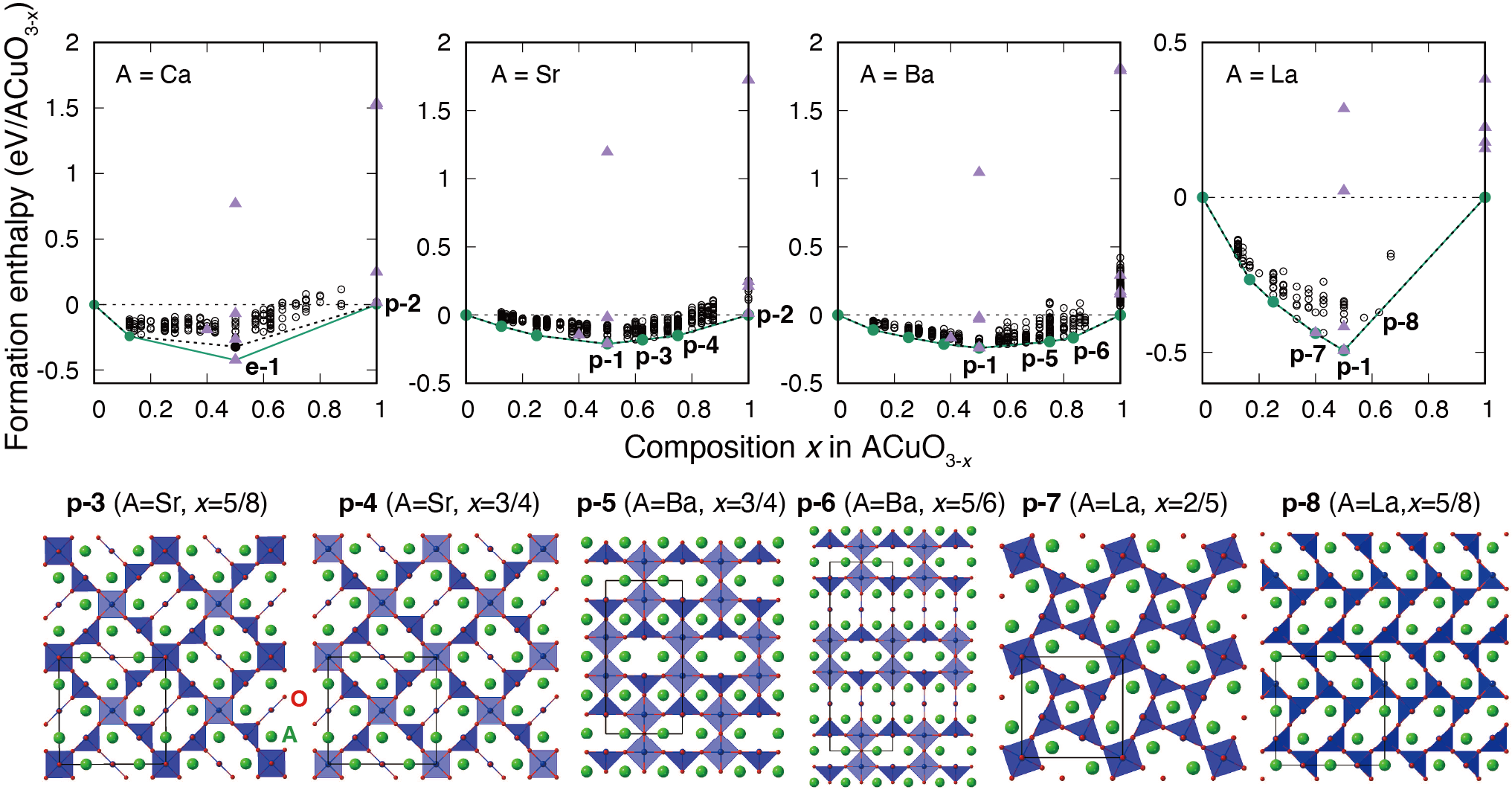
酸素欠損ペロブスカイトにおける第一原理計算とベイズ最適化による構造予測 [A. Seko and S. Ishiwata, PRB (2020)]
また、より大きな単位胞を使用した構造探索や、多成分系、複雑な化学組成を対象とする場合、構造列挙の数が膨大になります。こうした場合、効率的なデータ構造を活用することが非常に重要です。例えば、Decision diagramを活用することで、膨大な候補の中から必要な構造のみを効率よく抽出できます。 特に、多くの非等価な構造を含む構造集合から、解となりうる構造に絞り込むことが求められます。データ構造の形式を保持したままスクリーニングを行うことで、従来の方法よりもはるかに効率的に構造探索を進めることができます。 このような私たちが開発している機械学習やアルゴリズムを用いた効率的な構造探索手法は、新たな材料開発にも大いに貢献する可能性を秘めており、今後さらに多くの応用が期待されています。