高精度第一原理熱力学計算手法の開発¶
第一原理計算を材料研究に応用する試みは1980年代に始まり、計算機と計算技術の進歩により着実に成果を上げてきました。そして、21世紀に入るとさらに大きな進展があり、温度の効果を定量的に計算できるようになりました。これにより、物質の比熱や自由エネルギーといった熱力学関数を第一原理計算で求めることが可能となり、状態図、相転移、拡散など、材料科学の根幹をなす現象にアプローチできるようになりました。 当研究室では、酸化物セラミックス材料、半導体、金属材料において、すでに多くの先駆的な成果を報告しています。
第一原理熱力学計算では、典型的な第一原理計算と新しい技術を組み合わせることで、これまで実現できなかった熱力学計算を達成しています。例えば、格子振動の効果を求めるために、フォノンの状態密度やフォノン相互作用を第一原理計算や機械学習ポテンシャルを用いて計算しています。また、固溶体や化合物の置換型原子配列については、クラスター展開法やモンテカルロ法などの統計力学的手法を導入し、さらなる精度の向上を目指しています。 私たちは、このような新しい計算方法を積極的に材料科学の問題に応用し、革新的な解決策を提供しています。
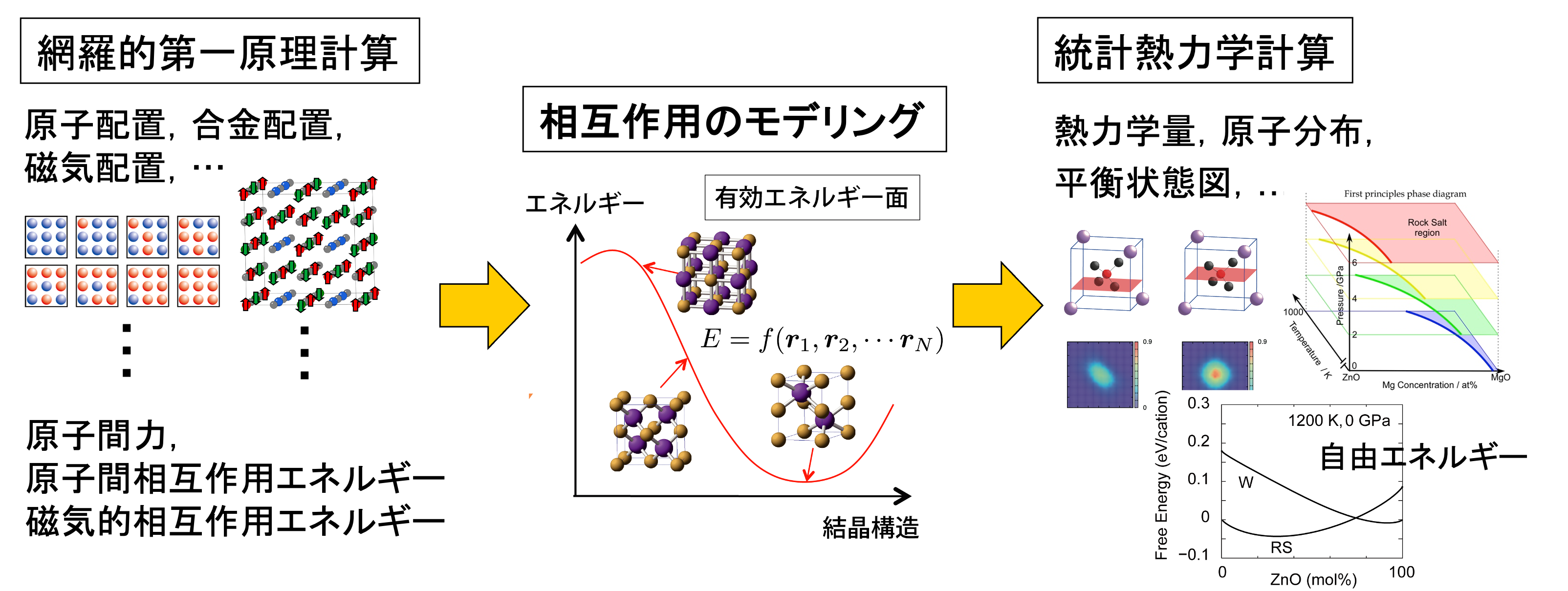
第一原理熱力学計算の一般的な流れ
複合酸化物や酸化物固溶体の構造予測・状態図計算
酸化物セラミックスやワイドギャップ半導体などの化合物系では、基本的な熱力学データが不足していることが多く、実験結果に基づいた状態図の報告例は限られています。また、第一原理計算を用いて計算状態図を作成するには、結晶構造が一般的に複雑であるため、従来の単純化された統計熱力学的手法が適用できないことや、第一原理計算そのものの精度に限界があることが課題となっていました。 近年、第一原理熱力学計算により、未知の複雑系における階層構造や状態図を予測できるようになっています。従来の実験的情報と第一原理計算による計算データを組み合わせることで、合理的かつ効率的な材料開発が進み、情報の正確さも向上しています。
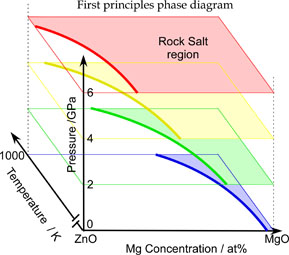
第一原理熱力学計算によるMgO-ZnO擬2元系状態図